Геномика
Геномика
Геномика - это междисциплинарная область биологии, в которой основное внимание уделяется структуре, функциям, эволюции, картированию и редактированию геномов. Геном - это полный набор ДНК организма, включая все его гены. В отличие от генетики, которая относится к изучению отдельных генов и их роли в наследовании, геномика направлена на коллективную характеристику и количественную оценку всех генов организма, их взаимосвязей. и влияние на организм. Гены могут управлять производством белков с помощью ферментов и молекул-посредников. В свою очередь, белки составляют структуры тела, такие как органы и ткани, а также контролируют химические реакции и передают сигналы между клетками. Геномика также включает в себя секвенирование и анализ геномов с использованием высокопроизводительного секвенирования ДНК и биоинформатики для сборки и анализа функции и структуры полных геномов. Достижения в области геномики вызвали революцию в исследованиях, основанных на открытиях, и в системной биологии, чтобы облегчить понимание даже самых сложных биологических систем, таких как мозг.
Эта область также включает исследования внутригеномных (внутри генома) явлений такие как эпистаз (влияние одного гена на другой), плейотропия (один ген влияет на более чем один признак), гетерозис (сила гибрида) и другие взаимодействия между локусами и аллелями в геноме.
Содержание
История
Этимология
От греческого ΓΕΝ gen , «ген» (гамма, эпсилон, ню, эпсилон), что означает «становиться, создать, сотворение, рождение "и последующие варианты: генеалогия, генезис, генетика, ген, геном, генотип, род и т. д. Слово геном (от немецкого Genom , приписываемый Гансу Винклеру) использовался в английском языке еще в 1926 году, термин геномика был придуман Томом Родериком, генетиком из лаборатории Джексона (Бар-Харбор, штат Мэн), за пивом на встрече, проведенной я n Мэриленд о картировании генома человека в 1986 году.
Первые попытки секвенирования
После подтверждения Розалиндой Франклин спиральной структуры ДНК Джеймс Д. Уотсон и Фрэнсис Крик опубликовали Структура ДНК в 1953 году и публикация Фредом Сэнгером аминокислотной последовательности инсулина в 1955 году, секвенирование нуклеиновых кислот стало основной целью ранних молекулярных биологов. В 1964 году Роберт В. Холли и его коллеги опубликовали первую когда-либо определенную последовательность нуклеиновой кислоты - рибонуклеотидную последовательность РНК-переносчика аланина. Продолжая эту работу, Маршалл Ниренберг и Филип Ледер раскрыли триплетную природу генетического кода и в своих экспериментах смогли определить последовательности 54 из 64 кодонов. В 1972 году Уолтер Фирс и его команда из Лаборатории молекулярной биологии Гентского университета (Гент, Бельгия) первыми определили последовательность гена: гена белка оболочки бактериофага MS2. Группа Файерса расширила свою работу с белками оболочки MS2, определив полную нуклеотидную последовательность MS2-РНК бактериофага (чей геном кодирует всего четыре гена в 3569 парах оснований) и вируса обезьяны 40 в 1976 и 1978 годах соответственно.
Разработана технология секвенирования ДНК
В дополнение к его плодотворной работе над аминокислотной последовательностью инсулина Фредерик Сэнгер и его коллеги сыграли ключевую роль в разработке методов секвенирования ДНК, которые позволили создать всеобъемлющие проекты секвенирования генома. В 1975 году он и Алан Коулсон опубликовали процедуру секвенирования с использованием ДНК-полимеразы с радиоактивно меченными нуклеотидами, которую он назвал техникой плюс и минус . Это включало два тесно связанных метода, которые генерировали короткие олигонуклеотиды с определенными 3'-концами. Их можно фракционировать с помощью электрофореза в полиакриламидном геле (так называемый электрофорез в полиакриламидном геле) и визуализировать с помощью авторадиографии. Эта процедура позволяла секвенировать до 80 нуклеотидов за один раз и была большим улучшением, но все же была очень трудоемкой. Тем не менее, в 1977 году его группа смогла секвенировать большую часть из 5386 нуклеотидов одноцепочечного бактериофага φX174, завершив первый полностью секвенированный геном на основе ДНК. Уточнение метода Плюс и Минус привело к обрыву цепи или методу Сенгера (см. Ниже), который лег в основу методов секвенирования ДНК, картирования генома, хранения данных и биоинформатического анализа. наиболее широко использовался в последующие четверть века исследований. В том же году Уолтер Гилберт и Аллан Максам из Гарвардского университета независимо друг от друга разработали метод Максама-Гилберта (также известный как химический метод ) секвенирования ДНК, предполагающий преимущественное расщепление ДНК по известным основаниям, менее эффективный метод. За свою новаторскую работу по секвенированию нуклеиновых кислот Гилберт и Сэнгер разделили половину Нобелевской премии по химии 1980 года с Полом Бергом (рекомбинантная ДНК).
Полные геномы
Появление этих технологий привело к быстрому увеличению масштабов и скорости выполнения проектов по секвенированию генома. О первой полной последовательности генома эукариотической органеллы, митохондрии человека (16568 п.н., около 16,6 т.п.н.), было сообщено в 1981 году, а первые геномы хлоропластов последовали в 1986 году. В 1992 году первая эукариотическая хромосома, хромосома III пивных дрожжей Saccharomyces cerevisiae (315 т.п.н.) секвенировали. Первым свободноживущим организмом, который был секвенирован, был Haemophilus influenzae (1,8 МБ) в 1995 году. В следующем году консорциум исследователей из лабораторий Северной Америки, Европы и Японии объявил о завершении первая полная последовательность генома эукариота, S. cerevisiae (12,1 МБ), и с тех пор секвенирование геномов продолжалось с экспоненциально растущей скоростью. По состоянию на октябрь 2011 года полные последовательности доступны для: 2719 вирусов, 1115 архей и бактерий и 36 эукариот, из которых около половины составляют грибы.
Большинство микроорганизмов, чьи геномы были полностью секвенированы, проблематичны. патогены, такие как Haemophilus influenzae , что привело к явному смещению их филогенетического распределения по сравнению с широтой микробного разнообразия. Из других секвенированных видов большинство было выбрано потому, что они были хорошо изученными модельными организмами или обещали стать хорошими моделями. Дрожжи ( Saccharomyces cerevisiae ) долгое время были важным модельным организмом для эукариотической клетки, в то время как плодовая мушка Drosophila melanogaster была очень важным инструментом (особенно на ранних этапах до-молекулярной генетика). Червь Caenorhabditis elegans - часто используемая простая модель для многоклеточных организмов. Рыбка данио Brachydanio rerio используется для многих исследований развития на молекулярном уровне, а растение Arabidopsis thaliana является модельным организмом для цветущих растений. Японский иглобрюх ( Takifugu rubripes ) и пятнистый зеленый иглобрюх ( Tetraodon nigroviridis ) интересны своими небольшими и компактными геномами, которые содержат очень мало некодирующей ДНК по сравнению с большинством видов. . Собака млекопитающих ( Canis familis ), коричневая крыса ( Rattus norvegicus ), мышь ( Mus musculus ) и шимпанзе ( Pan troglodytes ) являются важными модельными животными в медицинских исследованиях.
Черновой набросок генома человека был завершен проектом Human Genome Project в начале 2001 года, вызвав много шума. В рамках этого проекта, завершенного в 2003 году, был секвенирован весь геном одного конкретного человека, и к 2007 году эта последовательность была объявлена «завершенной» (менее одной ошибки на 20 000 оснований и собраны все хромосомы). За прошедшие с тех пор годы геномы многих других людей были секвенированы, частично под эгидой проекта 1000 Genomes Project, который объявил о секвенировании 1092 генома в октябре 2012 года. Завершение этого проекта стало возможным благодаря разработке значительно большего количества эффективные технологии секвенирования и потребовали выделения значительных ресурсов в области биоинформатики в результате большого международного сотрудничества. Постоянный анализ геномных данных человека имеет глубокие политические и социальные последствия для человеческих обществ.
Революция "омиков"
Англоязычный неологизм омикс неофициально относится к области исследований в биология, оканчивающаяся на -omics , например геномика, протеомика или метаболомика. Связанный суффикс -ome используется для обращения к объектам исследования таких областей, как геном, протеом или метаболом соответственно. Суффикс -ome , используемый в молекулярной биологии, относится к некоторой совокупности ; аналогично омикс стал относиться к изучению больших, всеобъемлющих наборов биологических данных. Хотя рост использования этого термина заставил некоторых ученых (среди прочих, Джонатан Эйзен) утверждать, что он был перепродан, он отражает изменение ориентации на количественный анализ полного или почти полного ассортимента всех составляющих система. Например, при изучении симбиозов исследователи, которые раньше ограничивались изучением одного генного продукта, теперь могут одновременно сравнивать общий набор нескольких типов биологических молекул.
Анализ генома
После выбора организма проекты генома включают три компонента: секвенирование ДНК, сборку этой последовательности для создания представления исходной хромосомы, а также аннотацию и анализ этого представления.
Секвенирование
Исторически секвенирование выполнялось в центрах секвенирования , централизованных объектах (от крупных независимых институтов, таких как Объединенный институт генома, секвенирующих десятки терабаз в год, до местных основных центров молекулярной биологии), где проводятся исследования лаборатории с дорогостоящим оборудованием и необходимой технической поддержкой. Однако по мере того, как технология секвенирования продолжает совершенствоваться, новое поколение эффективных настольных секвенаторов с быстрым оборотом стало доступным для средней академической лаборатории. В целом подходы к секвенированию генома делятся на две большие категории: дробовик и высокопроизводительное (или секвенирование следующего поколения ).
Секвенирование дробовиком - это метод секвенирования, разработанный для анализа последовательностей ДНК длиной более 1000 пар оснований, вплоть до целых хромосом. Он назван по аналогии с быстро расширяющейся квазислучайной схемой стрельбы из дробовика. Поскольку секвенирование с помощью гель-электрофореза можно использовать только для довольно коротких последовательностей (от 100 до 1000 пар оснований), более длинные последовательности ДНК должны быть разбиты на случайные небольшие сегменты, которые затем секвенируются для получения считываний . Множественные перекрывающиеся чтения целевой ДНК получают путем выполнения нескольких раундов этой фрагментации и секвенирования. Затем компьютерные программы используют перекрывающиеся концы разных операций чтения, чтобы собрать их в непрерывную последовательность. Секвенирование дробовиком - это случайный процесс выборки, требующий избыточной выборки, чтобы гарантировать, что данный нуклеотид представлен в реконструированной последовательности; среднее число считываний, при которых происходит избыточная выборка генома, называется охватом.
На протяжении большей части своей истории технология, лежащая в основе секвенирования дробовика, была классическим методом обрыва цепи или «методом Сэнгера», который основан на избирательном включении дидезоксинуклеотидов, завершающих цепь, ДНК-полимеразой во время репликации ДНК in vitro. В последнее время секвенирование методом дробовика было вытеснено высокопроизводительными методами секвенирования, особенно для крупномасштабных автоматизированных анализов генома. Однако метод Сэнгера по-прежнему широко используется, в первую очередь для небольших проектов и для получения особо длинных считываний непрерывных последовательностей ДНК (> 500 нуклеотидов). Для методов терминации цепи требуются одноцепочечная ДНК-матрица, ДНК-праймер, ДНК-полимераза, нормальные дезоксинуклеозидтрифосфаты (dNTP) и модифицированные нуклеотиды (дидезоксиNTP), которые останавливают удлинение цепи ДНК. В этих обрывающих цепь нуклеотидах отсутствует 3'-ОН группа, необходимая для образования фосфодиэфирной связи между двумя нуклеотидами, из-за чего ДНК-полимераза прекращает удлинение ДНК при включении ddNTP. ДдНТФ могут быть помечены радиоактивно или флуоресцентно для обнаружения в секвенаторах ДНК. Как правило, эти машины могут секвенировать до 96 образцов ДНК за одну серию (прогон) за 48 прогонов в день.
Высокий спрос на дешевое секвенирование стимулировал развитие высокопроизводительного секвенирования технологии, которые распараллеливают процесс секвенирования, создавая тысячи или миллионы последовательностей одновременно. Высокопроизводительное секвенирование предназначено для снижения стоимости секвенирования ДНК по сравнению со стандартными методами определения терминатора с использованием красителя. При сверхвысокопроизводительном секвенировании можно параллельно выполнять до 500 000 операций секвенирования путем синтеза.
Метод секвенирования красителей Illumina основан на использовании обратимых терминаторов красителей и был разработан в 1996 г. Женевский институт биомедицинских исследований, Паскаль Майер и Лоран Фаринелли. В этом методе молекулы ДНК и праймеры сначала прикрепляются к слайду и амплифицируются с помощью полимеразы, так что образуются локальные клональные колонии, первоначально названные «колониями ДНК». Для определения последовательности добавляют четыре типа оснований обратимых терминаторов (RT-основания) и смывают невключенные нуклеотиды. В отличие от пиросеквенирования, цепи ДНК удлиняются на один нуклеотид за раз, и получение изображений может выполняться с задержкой, что позволяет захватывать очень большие массивы колоний ДНК с помощью последовательных изображений, полученных с одной камеры. Разделение ферментативной реакции и захвата изображения обеспечивает оптимальную производительность и теоретически неограниченную емкость секвенирования; при оптимальной конфигурации конечная производительность прибора зависит только от скорости аналого-цифрового преобразования камеры. Камера делает снимки флуоресцентно меченных нуклеотидов, затем краситель вместе с концевым блокатором 3 'химически удаляется из ДНК, обеспечивая следующий цикл.
Альтернативный подход - ионно-полупроводниковое секвенирование - основан на стандартной химии репликации ДНК. Эта технология измеряет выделение иона водорода каждый раз, когда вводится основание. Микролунка, содержащая матричную ДНК, заполняется одним нуклеотидом, если нуклеотид комплементарен матричной цепи, он будет включен, и ион водорода будет высвобожден. Этот выпуск запускает ионный датчик ISFET. Если в матричной последовательности присутствует гомополимер, несколько нуклеотидов будут включены в один цикл наводнения, и обнаруженный электрический сигнал будет пропорционально выше.
Сборка
Сборка последовательности относится к выравниванию и слияние фрагментов гораздо более длинной последовательности ДНК, чтобы восстановить исходную последовательность. Это необходимо, поскольку текущая технология секвенирования ДНК не может считывать целые геномы как непрерывную последовательность, а скорее считывает небольшие фрагменты от 20 до 1000 оснований, в зависимости от используемой технологии. Технологии секвенирования третьего поколения, такие как PacBio или Oxford Nanopore, обычно генерируют чтения секвенирования длиной & gt; 10 kb; однако у них высокий уровень ошибок - примерно 15 процентов. Обычно короткие фрагменты, называемые чтениями, являются результатом дробного секвенирования геномной ДНК или транскриптов генов (EST).
Сборку можно в целом разделить на два подхода: de novo сборка, для геномы, которые не похожи ни на одну секвенированную в прошлом, и сравнительную сборку, при которой в качестве эталона во время сборки используется существующая последовательность близкородственного организма. По сравнению со сравнительной сборкой, сборка de novo сложна в вычислительном отношении (NP-hard), что делает ее менее подходящей для технологий NGS с коротким чтением. В рамках парадигмы сборки de novo существуют две основные стратегии сборки: стратегии эйлерова пути и стратегии перекрытия-компоновки-консенсуса (OLC). Стратегии OLC в конечном итоге пытаются создать гамильтонов путь через граф перекрытия, что является NP-сложной проблемой. Стратегии эйлерова пути вычислительно более управляемы, потому что они пытаются найти эйлеров путь через граф де Брюйна.
Завершенные геномы определяются как имеющие единственную непрерывную последовательность без неоднозначностей, представляющих каждый репликон.
Аннотация
Сборка последовательности ДНК сама по себе не имеет большого значения без дополнительного анализа. Аннотации генома - это процесс присоединения биологической информации к последовательностям, который состоит из трех основных этапов:
- идентификация частей генома, которые не кодируют белки;
- идентификация элементов на геном, процесс, называемый предсказанием генов, и
- привязка биологической информации к этим элементам.
Инструменты автоматического аннотирования пытаются выполнить эти шаги in silico , в отличие от ручного аннотирования (также известного как курирование), который требует человеческого опыта и потенциальной экспериментальной проверки. В идеале эти подходы сосуществуют и дополняют друг друга в одном конвейере аннотаций (также см. Ниже).
Традиционно базовым уровнем аннотации является использование BLAST для поиска сходства, а затем аннотирование геномов на основе гомологов . Совсем недавно на платформу аннотаций добавлена дополнительная информация. Дополнительная информация позволяет ручным аннотаторам деконволютировать несоответствия между генами, которым даны одинаковые аннотации. Некоторые базы данных используют контекстную информацию генома, оценки сходства, экспериментальные данные и интеграции других ресурсов для предоставления аннотаций генома с помощью своего подхода к подсистемам. Другие базы данных (например, Ensembl) полагаются как на тщательно отобранные источники данных, так и на ряд программных инструментов в своем конвейере автоматизированной аннотации генома. Структурная аннотация состоит из идентификации геномных элементов, в первую очередь ORF, и их локализации или структуры гена. Функциональная аннотация состоит из присоединения биологической информации к геномным элементам.
Конвейеры секвенирования и базы данных
Необходимость воспроизводимости и эффективного управления большим объемом связанных данных с проектами генома означают, что вычислительные конвейеры имеют важное применение в геномике.
Области исследований
Функциональная геномика
Функциональная геномика - это область молекулярной биологии, в которой делается попытка использовать огромное количество данных, полученных в ходе геномных проектов (таких как проекты секвенирования генома), для описания функций и взаимодействий генов (и белков). Функциональная геномика фокусируется на динамических аспектах, таких как транскрипция генов, трансляция и белок-белковые взаимодействия, в отличие от статических аспектов геномной информации, таких как последовательность или структуры ДНК. Функциональная геномика пытается ответить на вопросы о функции ДНК на уровне генов, транскриптов РНК и белковых продуктов. Ключевой характеристикой исследований функциональной геномики является их общегеномный подход к этим вопросам, как правило, с использованием высокопроизводительных методов, а не более традиционного подхода «ген за геном».
Основным разделом геномики является все еще занимается секвенированием геномов различных организмов, но знание полных геномов создало возможность для области функциональной геномики, в основном связанной с паттернами экспрессии генов в различных условиях. Наиболее важными инструментами здесь являются микрочипы и биоинформатика.
Структурная геномика
Структурная геномика стремится описать трехмерную структуру каждого белка, кодируемого данным геномом. Этот геномный подход позволяет использовать высокопроизводительный метод определения структуры путем сочетания экспериментального и модельного подходов. Принципиальное различие между структурной геномикой и традиционным структурным предсказанием состоит в том, что структурная геномика пытается определить структуру каждого белка, кодируемого геномом, а не сосредотачиваться на одном конкретном белке. При наличии полногеномных последовательностей предсказание структуры может быть выполнено быстрее с помощью комбинации экспериментального и модельного подходов, особенно потому, что наличие большого количества секвенированных геномов и ранее решенных белковых структур позволяет ученым моделировать структуру белка на структурах ранее решенных. гомологи. Структурная геномика включает использование большого количества подходов к определению структуры, включая экспериментальные методы с использованием геномных последовательностей или подходы, основанные на моделировании, основанные на последовательности или структурной гомологии с белком известной структуры или основанные на химических и физических принципах для белка, не имеющего гомологии с белком. любая известная структура. В отличие от традиционной структурной биологии, определение структуры белка с помощью структурной геномики часто (но не всегда) происходит до того, как что-либо известно о функции белка. Это ставит новые задачи в структурной биоинформатике, т. Е. В определении функции белка по его трехмерной структуре.
Эпигеномика
Эпигеномика - это исследование полного набора эпигенетических модификаций генетического материала клетки. , известный как эпигеном. Эпигенетические модификации - это обратимые модификации клеточной ДНК или гистонов, которые влияют на экспрессию генов без изменения последовательности ДНК (Russell 2010, стр. 475). Двумя наиболее характерными эпигенетическими модификациями являются метилирование ДНК и модификация гистонов. Эпигенетические модификации играют важную роль в экспрессии и регуляции генов и участвуют во многих клеточных процессах, таких как дифференцировка / развитие и туморогенез. Изучение эпигенетики на глобальном уровне стало возможным только недавно благодаря адаптации геномных высокопроизводительных анализов.
Метагеномика
Метагеномика - это изучение метагеномов , генетический материал, извлеченный непосредственно из образцов окружающей среды. Широкая область может также называться экологической геномикой, экогеномикой или геномикой сообщества. В то время как традиционная микробиология и секвенирование микробного генома основаны на культивируемых клональных культурах, раннее секвенирование генов окружающей среды клонирует определенные гены (часто ген 16S рРНК) для получения профиля разнообразия в естественном образце. Такая работа показала, что подавляющее большинство микробного биоразнообразия было упущено методами культивирования. В недавних исследованиях используется «дробное» секвенирование по Сэнгеру или массивно-параллельное пиросеквенирование, чтобы получить в значительной степени объективные образцы всех генов от всех членов выбранных сообществ. Благодаря своей способности раскрывать ранее скрытое разнообразие микроскопической жизни, метагеномика предлагает мощную линзу для наблюдения за микробным миром, которая может революционизировать понимание всего живого мира.
Модельные системы
Бактериофаги играли и продолжают играть ключевую роль в бактериальной генетике и молекулярной биологии. Исторически они использовались для определения структуры генов и регуляции генов. Также первым секвенированным геномом был бактериофаг. Однако исследования бактериофагов не привели к революции в геномике, в которой явно преобладает бактериальная геномика. Лишь совсем недавно изучение геномов бактериофагов стало популярным, что позволило исследователям понять механизмы, лежащие в основе эволюции фагов. Последовательности генома бактериофага могут быть получены путем прямого секвенирования изолированных бактериофагов, но также могут быть получены как часть микробных геномов. Анализ бактериальных геномов показал, что значительная часть микробной ДНК состоит из профаговых последовательностей и профагоподобных элементов. Подробный анализ этих последовательностей в базе данных позволяет понять роль профагов в формировании бактериального генома: в целом, этот метод подтвердил многие известные группы бактериофагов, что делает его полезным инструментом для прогнозирования взаимосвязи профагов с бактериальными геномами.
В настоящее время существует 24 цианобактерии, для которых доступна полная последовательность генома. 15 из этих цианобактерий происходят из морской среды. Это шесть штаммов Prochlorococcus , семь морских штаммов Synechococcus , Trichodesmium erythraeum IMS101 и Crocosphaera watsonii WH8501. Несколько исследований продемонстрировали, как эти последовательности могут быть очень успешно использованы для вывода важных экологических и физиологических характеристик морских цианобактерий. Однако в настоящее время ведется еще много проектов по геному, среди которых есть еще изоляты Prochlorococcus и морских Synechococcus , Acaryochloris и Prochloron. , N2-фиксирующие нитчатые цианобактерии Nodularia spumigena , Lyngbya aestuarii и Lyngbya majuscula , а также бактериофаги, заражающие морские цианобактерии. Таким образом, растущий объем информации о геноме можно использовать и в более общем плане для решения глобальных проблем, применяя сравнительный подход. Некоторыми новыми и захватывающими примерами прогресса в этой области являются идентификация генов регуляторных РНК, понимание эволюционного происхождения фотосинтеза или оценка вклада горизонтального переноса генов в анализируемые геномы.
Приложения геномики
Геномика нашла применение во многих областях, включая медицину, биотехнологию, антропологию и другие социальные науки.
Геномная медицина
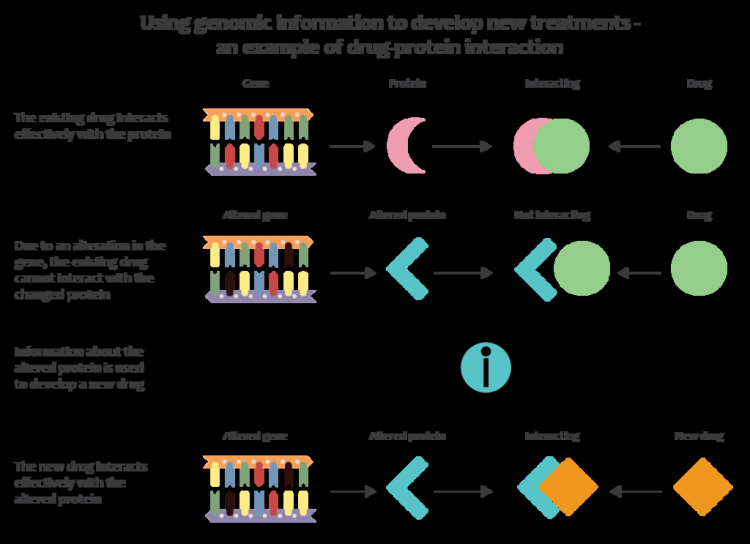
Геномная медицина следующего поколения Технологии позволяют клиницистам и биомедицинским исследователям резко увеличить объем геномных данных, собираемых в больших исследуемых популяциях. В сочетании с новыми информатическими подходами, которые объединяют многие виды данных с геномными данными в исследованиях болезней, это позволяет исследователям лучше понять генетические основы реакции на лекарства и болезни. Ранние попытки применить геном в медицине были предприняты командой Стэнфорда во главе с Юаном Эшли, который разработал первые инструменты для медицинской интерпретации генома человека. Например, исследовательская программа Все мы направлена на сбор данных о последовательности генома от 1 миллиона участников, чтобы стать важным компонентом исследовательской платформы точной медицины.
Синтетическая биология и биоинженерия
Рост знаний в области геномики позволил использовать все более сложные приложения синтетической биологии. В 2010 году исследователи из Института Дж. Крейга Вентера объявили о создании частично синтетического вида бактерий, Mycoplasma labratorium , производного от генома Mycoplasma genitalium .
Геномика сохранения
Специалисты по охране природы могут использовать информацию, собранную путем геномного секвенирования, для более точной оценки генетических факторов, имеющих ключевое значение для сохранения видов, таких как генетическое разнообразие популяции или гетерозиготность индивида для рецессивного наследственное генетическое заболевание. Используя геномные данные для оценки эффектов эволюционных процессов и выявления закономерностей в вариациях в данной популяции, защитники природы могут сформулировать планы по оказанию помощи данному виду без того количества переменных, которые остаются неизвестными, чем те, которые не учитываются стандартными генетическими подходами.

Генетика может не иметь большого отношения к продолжительности вашей жизни, но это имеет значение
Согласно новому исследованию, продолжительность нашей жизни меньше связана с …

Генриетта Лакс: что нужно знать о ее «бессмертных» клетках и почему ее история является примером расизма в медицине
Генриетта Лакс и ее «бессмертные» клетки были неотъемлемой частью сообщества …
